
Hasta
50%
de probabilidad de transmitir la patología por herencia dominante
En marcha
2
proyectos de investigación sobre nuevos genes implicados
Más de
100
genes implicados y más de 2.000 mutaciones identificadas
¿Qué es la retinosis pigmentaria?
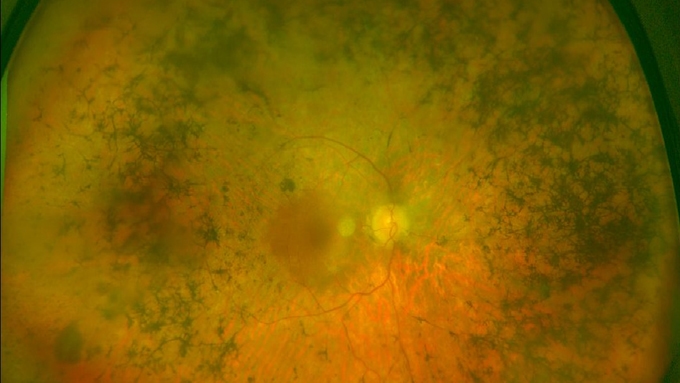
Es una patología hereditaria que se caracteriza por la degeneración precoz y progresiva de las células retinianas, fundamentalmente, las fotorreceptoras: los bastones (visión nocturna) y los conos (percepción de colores y detalles), que convierten la luz en señales que se transmiten al cerebro.
La retinosis pigmentaria se engloba dentro del grupo de las enfermedades consideradas “raras” o minoritarias –afecta aproximadamente a 1 de cada 3.000 personas–, aunque se trata de la distrofia hereditaria de la retina más frecuente.
¿Por qué se produce?
La retinosis pigmentaria es una enfermedad genética de la retina.
Actualmente, hay descritos más de 100 genes implicados y se han identificado más de 2.000 mutaciones. Sin embargo, estas solo explican una parte de los casos de retinosis pigmentaria y, en alrededor de un 25-30% de las familias afectadas a las que se realiza un diagnóstico genético, no se logra identificar la causa molecular. Esto significa que todavía quedan nuevos genes y mutaciones por asociar a la enfermedad, un reto al que se orienta la investigación y para el que Fundación IMO tiene actualmente 2 proyectos en marcha en el laboratorio.
Herencia de la retinosis pigmentaria
La genética es compleja en esta distrofia de la retina, ya que puede transmitirse a través de diferentes patrones de herencia:
- Dominante: afectos en todas las generaciones de la familia.
- Recesiva: salta generaciones. Hay afectos en una generación y, en las otras, miembros portadores.
- Ligada al cromosoma X: afectos solo varones y mujeres portadoras.
¿Cómo se puede prevenir?

Dado que es de origen genético, no se puede evitar la aparición de la retinosis pigmentaria. Sin embargo, detectarla tempranamente permite anticiparse a la enfermedad y realizar un buen seguimiento oftalmológico de los pacientes para controlar posibles problemas asociados (por ejemplo, otras alteraciones en la retina o catarata).
Como refuerzo del diagnóstico clínico, contar con un diagnóstico genético que determine la causa molecular de la patología es de utilidad para identificar su patrón de herencia e indicar la probabilidad de transmitirla. Además, permite alertar a familiares portadores y a miembros afectos que aún no la han desarrollado, pero que pueden manifestarla en el futuro.
En familias con antecedentes de la enfermedad, existe la posibilidad de hacer un diagnóstico genético preimplantacional para, mediante fecundación in vitro, transferir al útero materno embriones libres de la mutación. Por otro lado, el diagnóstico genético prenatal, a partir de una muestra de líquido amniótico obtenida durante el embarazo, permite detectar antes del nacimiento la presencia de la mutación responsable de la retinosis pigmentaria.
Síntomas
Aunque la mutación genética está presente desde el nacimiento, la retinosis pigmentaria suele comenzar a dar los primeros síntomas en la adolescencia. La pérdida paulatina de visión que provoca afecta generalmente a ambos ojos, aunque de forma desigual, y acostumbra a ser lenta. Ahora bien, la velocidad de progresión de la patología y su severidad es muy variable, en función de la mutación y del gen afectado en cada caso.
Estos son algunos de los principales síntomas que experimentan los pacientes:
- Ceguera nocturna: la dificultad de visión en entornos poco iluminados, así como la mala adaptación de la claridad a la oscuridad, es una de las primeras manifestaciones de la retinosis pigmentaria.
- Visión en túnel: la reducción del campo visual hace que los pacientes vayan perdiendo visión periférica, por lo que es habitual que sufran golpes y caídas frecuentes o que tengan problemas para localizar y coger objetos a su alrededor. Por el contrario, la visión central se mantiene hasta fases tardías de la enfermedad.
- Deslumbramientos: la percepción de destellos y “flashes”, sobre todo en condiciones de mucha luminosidad, resulta muy molesta y hace necesario el uso de gafas de sol con filtros especiales.
- Disminución de la agudeza visual: la capacidad de ver con nitidez y distinguir detalles se merma cuando la patología está evolucionada.
- Alteración en la percepción de los colores: también ocurre en casos avanzados de retinosis pigmentaria.
Tratamientos asociados
Actualmente, no es posible detener la pérdida de visión que provoca la retinosis pigmentaria (aunque sí puede aprovecharse el resto visual de los pacientes gracias a ayudas de baja visión).
Para dar con un tratamiento efectivo, es una de las patologías oculares de origen hereditario sobre las que más se está investigando, con varias líneas de estudio que esperan obtener sus frutos en los próximos años:
Terapias génicas: están en fase avanzada de investigación y consisten en el reemplazo del gen defectuoso por uno sano para evitar que progrese la enfermedad. Requieren conocer la causa molecular en cada paciente, por lo que solo son aptas para aquellos a quienes previamente se haya identificado la mutación en el gen responsable a través del diagnóstico genético, imprescindible para poder diseñar una estrategia terapéutica individualizada.
Terapias celulares: basadas en la introducción de células sanas en el tejido retiniano afectado, al que deben integrarse (cumpliendo con las complejas funciones de las células de la retina después de haber sido cultivadas en el laboratorio a partir de las células madre obtenidas de otro tejido). Esta estrategia es independiente de la causa molecular y, una vez logren superarse las dificultades del proceso, podrá aplicarse en estadios más severos de la patología que las terapias génicas.
Chip de retina: la visión biónica ya hace posible la percepción de estímulos luminosos para facilitar la localización e identificación de objetos en personas invidentes. Se está trabajando en modelos que aporten cada vez una mayor resolución de las imágenes y que permitan dar el salto definitivo de esta tecnología que estimula artificialmente la retina.
Los especialistas de IMO están involucrados tanto en proyectos de investigación que se llevan a cabo en el laboratorio de biología molecular para sentar las bases de la aplicación de terapias génicas y celulares, como en el desarrollo de la visión artificial. En este sentido, el Instituto ha implantado el chip de retina IRIS®II, por primera vez fuera de estudios en Europa, en una paciente con retinosis pigmentaria, además de seguir involucrado en nuevos modelos y avances.
Especialistas que tratan esta patología








Preguntas frecuentes
La retinitis pigmentaria es la enfermedad hereditaria más frecuente de la retina. Se caracteriza por una degeneración progresiva de la retina. Actualmente carece de tratamiento, y su gravedad hacen que sea una de las patologías oculares de origen genético sobre las que más se está investigando. La terapia génica está ofreciendo resultados muy esperanzadores. Aunque no existe todavía tratamiento, los pacientes con retinosis podrían acudir al Departamento de Genética de IMO, para llevar a cabo los estudios que están en proceso. Según se desprende de las conclusiones del Congreso internacional de Retina celebrado en IMO, estudios en fase I en humanos ya parecen estar demostrando cómo el uso de células madre para reemplazar células dañadas de la retina logra mejorar la agudeza visual de los pacientes. Esta terapia se aplica en pacientes que pierden células fotorreceptoras y/o del epitelio pigmentario, un tipo de células que no se regeneran, y que son fundamentales para la visión. Lo que se está consiguiendo con las nuevas terapias es reemplazarlas por células madre embrionarias o pluripotenciales extraídas de la piel o de otras partes del ojo, que, tras ser alteradas, son capaces de desarrollar la misma función que las células retinianas dañadas. En estos momentos, este tratamiento se está aplicando en fase de pruebas y con muy buenos resultados a pacientes con distrofias retinianas, retinosis pigmentaria y DMAE.
Es una técnica diagnóstica para determinar las estructuras patológicas y anormales que tiene la retina, tanto en sus vasos como en sus diferentes capas. Es válida para la degeneración de la mácula, para la retinopatía diabética y para otras muchas alteraciones maculares y para vasculopatías.
La angiografía es una técnica que sirve para delinear los casos de la retina o la coroides. Se utilizan diferentes contrastes, normalmente la fluoresceína sódica o el verde indocianina. Esta exploración también es muy útil para el diagnóstico de otras afecciones de la retina, especialmente del llamado epitelio pigmentado. En general, las angiografías sirven para estudiar múltiples enfermedades de la retina y su diagnóstico.
Aunque la terapia con células madre aún se encuentra en investigación, ya se está aplicando en fase de pruebas y con muy buenos resultados a pacientes con distrofias retinianas, retinosis pigmentaria y DMAE. Estudios en fase I en humanos parecen demostrar que el uso de células madre embrionarias o pluripotenciales logra mejorar la agudeza visual de los pacientes, ya que permite reemplazar las células fotorreceptoras y/o del epitelio pigmentario que no se regeneran y son fundamentales para la visión. Este resultado se consigue modificando genéticamente las células madre, extraídas de la piel u otras partes del ojo, para que puedan desarrollar la misma función que las células dañadas.
La DMAE (degeneración macular asociada a la edad) es uno de los grandes retos de la oftalmología en la actualidad. Sabemos que existen dos tipos: la forma seca y la forma húmeda. La seca es la que padecen los pacientes que, lentamente, pierden visión. Se ha demostrado que con tratamientos con antioxidantes y vitaminas se puede reducir esta pérdida visual, pese a que el detenimiento de la evolución no es muy espectacular. Las formas húmedas, llamadas así porque se produce líquido en la mácula, son las más destructivas, y el tratamiento actual combina la terapia fotodinámica, que se empezó a aplicar hace unos años, con otros tratamientos, lo que parece demostrar la obtención de efectos más positivos. Cada año, aparecen nuevas opciones para intentar avanzar en la lucha contra esta enfermedad.
Quizá te interese
IMO Grupo Miranza Barcelona
Josep María Lladó, 3
08035 Barcelona
Tel: 934 000 700
E-mail: informacion@imo.es
Ver mapa en Google Maps
En coche
Coordenadas navegador GPS:
41º 24’ 38” N – 02º 07’ 29” E
Salida 7 de la Ronda de Dalt (lado montaña). La clínica cuenta con un aparcamiento de más de 200 plazas.
En bus
Autobús H2: Rotonda de Bellesguard, parada 1540
Autobús 196: Josep Maria Lladó-Bellesguard, parada 3191
Autobuses H2, 123, 196: Ronda de Dalt – Bellesguard, parada 0071
Cómo llegar a IMO desde:
IMO Grupo Miranza Madrid
C/ Valle de Pinares Llanos, 3
28035 Madrid
Tel: 910 783 783
E-mail: administracion.madrid@imo.es
Transporte público
Metro Lacoma (línea 7)
Autobuses:
- Líneas 49 y 64, parada “Senda del Infante”
- Línea N21, parada «Metro Lacoma»
Horarios
Atención al paciente: de lunes a viernes, de 8 h a 21 h
IMO Grupo Miranza Andorra
Av. de les Nacions Unides, 17
AD700 Escaldes-Engordany, Andorra
Tel: (+376) 88 55 44
Ver mapa en Google Maps