Afectan a menos de
1
de cada 2.000 personas
Se conocen más de
250
genes relacionados
¿Qué son las distrofias de la retina?
Las distrofias retinianas son un conjunto heterogéneo de enfermedades hereditarias que provocan una pérdida progresiva y severa de visión, dado que alteran la anatomía y/o la función de la retina. Actualmente no tienen cura, aunque se está investigando para poder tratarlas, en los próximos años, con terapias génicas y celulares.
Estas patologías pueden causar un daño en las células fotorreceptoras, bien sean, predominantemente, los conos (responsables de la visión en detalle y de color), los bastones (especializados en la visión nocturna y periférica) o ambos a la vez. Es el caso de la enfermedad de Stargardt, la retinosis pigmentaria o la distrofia de conos-bastones, respectivamente.
También hay ciertas distrofias hereditarias, como la retinosquisis juvenil, la vitreoretinopatía exudativa familiar o el síndrome de Stickler, en las que se producen alteraciones del vítreo y de la retina. En otras, como la coroideremia, el problema de base está en la coroides, capa situada por debajo de la retina.
La mayoría de distrofias de retina son enfermedades localizadas exclusivamente en el ojo, aunque a veces pueden asociarse a manifestaciones extraoculares (síndrome de Usher, síndrome de Bardet-Biedl) en cuyo caso hablamos de distrofias retinianas sindrómicas.
Por su baja prevalencia, las distrofias de la retina son consideradas enfermedades minoritarias o “raras” (afectan a menos de 1 de cada 2.000 personas).
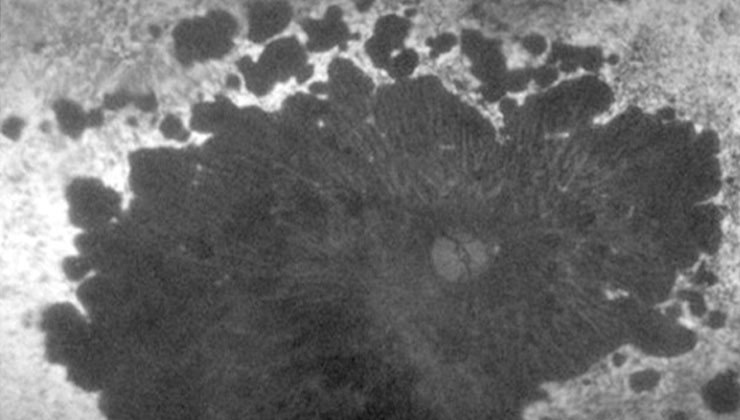
Hay varios estudios en marcha para poder ofrecer tratamiento a las distrofias de la retina. Esta imagen, igual que la superior, corresponde a una distrofia de conos-bastones, que afecta tanto a la visión central como periférica.
¿Por qué se producen?
Las distrofias de la retina tienen origen genético, por lo que pueden transmitirse de generación en generación mediante diferentes tipos de herencia.
- Herencia dominante: suelen haber afectados en todas las generaciones de la familia, ya que las personas portadoras de la mutación responsable de la enfermedad presentan la patología. Esta se transmite aproximadamente al 50% de los descendientes, como la enfermedad de Best.
- Herencia recesiva: habitualmente solo hay familiares afectos en una generación, ya que los pacientes portadores de una mutación están sanos, mientras que los portadores de dos mutaciones en el mismo gen desarrollan la enfermedad. Así ocurre en la enfermedad de Stargardt.
- Herencia ligada al cromosoma X: únicamente los hombres de la familia padecen la patología, aunque las mujeres pueden ser portadoras de la mutación y transmitir la enfermedad a sus hijos varones con un 50% de probabilidad. En este grupo se engloba, entre otras, la coroideremia.
La retinosis pigmentaria, la distrofia de la retina más frecuente, es un ejemplo de patología que puede transmitirse a través de los tres patrones de herencia mencionados, en función del gen implicado.
La genética de las distrofias de retina es compleja: una misma patología puede estar causada por varios genes y, a la vez, un mismo gen puede estar relacionado con diferentes enfermedades. Actualmente, se han descrito más de 250 genes asociados a las distrofias retinianas, aunque se estima que todavía faltan muchos por identificar.
¿Cómo se pueden prevenir?
La información genética contenida en el ADN de cada persona es la que determina que se desarrolle una distrofia de la retina y, por tanto, estas patologías hereditarias no se pueden prevenir. Sin embargo, un aspecto en el que sí se puede incidir es en su detección precoz por parte de oftalmólogos especialistas, mediante una revisión ocular completa que incluya una buena anamnesis y una exploración exhaustiva del fondo de ojo, además de técnicas complementarias, como la OCT, la autofluorescencia o las pruebas electrofisiológicas.
A menudo, las distrofias retinianas no son evidentes hasta fases avanzadas, cuando sus síntomas se hacen patentes. Para poder anticiparse a la enfermedad y predecir su evolución, es de gran utilidad conocer la causa molecular que la desencadena en cada paciente a través del diagnóstico genético, que también permite confirmar el diagnóstico clínico, identificar el patrón de herencia y ofrecer consejo genético a la familia, indicando la probabilidad de transmitir la patología y alertando a los familiares portadores.
Asimismo, el diagnóstico genético abre la puerta a prevenir, detener o revertir en el futuro la pérdida de visión de los pacientes afectados, mediante el desarrollo de nuevas terapias individualizadas que están en investigación.
Síntomas
Los síntomas, así como la rapidez de su evolución, varían según el tipo de distrofia de retina y cada paciente. No obstante, los más comunes son:
- Pérdida de agudeza visual: dificultad para apreciar detalles y realizar tareas de precisión.
- Reducción del campo de visión: visión “en túnel” o aparición de “puntos ciegos” (escotomas).
- Ceguera nocturna: problemas para ver de noche o en ambientes oscuros y mala adaptación a situaciones de poca luminosidad.
- Fotofobia y deslumbramientos: especiales molestias ante la luz y visión de “flashes” o reflejos en condiciones de alta luminosidad. El paso de un ambiente claro a un entorno oscuro –y viceversa– también genera problemas.
Otras posibles señales pueden ser la percepción deformada de objetos (metamorfopsia) o la alteración en la percepción de los colores (discromatopsia).
Tratamientos asociados
Las distrofias de la retina actualmente no tienen cura, debido a la dificultad de regenerar las células retinianas afectadas. El futuro tratamiento de estas patologías pasa por el diseño y aplicación de nuevas terapias génicas y celulares que permitan devolver visión o frenar su pérdida.
Actualmente, se están dando pasos importantes en esta línea de investigación (desde 2018, se ha empezado a comercializar la primera terapia génica en Estados Unidos, diseñada para un gen responsable de amaurosis congénita de Leber) y el Departamento de Genética del Instituto también se orienta a ello con varios proyectos promovidos por Fundación IMO.
Para preparar a los candidatos a estas terapias génicas es fundamental disponer de un diagnóstico genético, que IMO ofrece de forma pionera.
En paralelo, el Instituto también apuesta por otra vía alternativa y complementaria de tratamiento: la visión artificial, a cuyo desarrollo contribuye participando en estudios con nuevos modelos de chip de retina. Es el caso del IRIS®II, que ha implantado en la primera paciente de Europa.
Especialistas que tratan esta patología








Preguntas frecuentes
La retinitis pigmentaria es la enfermedad hereditaria más frecuente de la retina. Se caracteriza por una degeneración progresiva de la retina. Actualmente carece de tratamiento, y su gravedad hacen que sea una de las patologías oculares de origen genético sobre las que más se está investigando. La terapia génica está ofreciendo resultados muy esperanzadores. Aunque no existe todavía tratamiento, los pacientes con retinosis podrían acudir al Departamento de Genética de IMO, para llevar a cabo los estudios que están en proceso. Según se desprende de las conclusiones del Congreso internacional de Retina celebrado en IMO, estudios en fase I en humanos ya parecen estar demostrando cómo el uso de células madre para reemplazar células dañadas de la retina logra mejorar la agudeza visual de los pacientes. Esta terapia se aplica en pacientes que pierden células fotorreceptoras y/o del epitelio pigmentario, un tipo de células que no se regeneran, y que son fundamentales para la visión. Lo que se está consiguiendo con las nuevas terapias es reemplazarlas por células madre embrionarias o pluripotenciales extraídas de la piel o de otras partes del ojo, que, tras ser alteradas, son capaces de desarrollar la misma función que las células retinianas dañadas. En estos momentos, este tratamiento se está aplicando en fase de pruebas y con muy buenos resultados a pacientes con distrofias retinianas, retinosis pigmentaria y DMAE.
Existen muchas enfermedades oculares que están ligadas a la herencia cromosómica y está incluso determinado su tipo genético de defecto. Enfermedades de todas las partes del ojo pueden tener carácter hereditario. La más conocida es la retinosis pigmentaria.
Cuando la ptosis afecta a la visión o provoca molestias funcionales o psicológicas, el momento adecuado es cuanto antes. En niños, es esencial intervenir a tiempo si existe riesgo de ambliopía. En adultos, la decisión se toma tras una evaluación médica personalizada, teniendo en cuenta las necesidades del paciente y los problemas visuales o estéticos que presente.
Si no tiene gas ni aceite de silicona puede dormir en cualquier posición. Al no existir ningún elemento taponador (gas o aceite de silicona) no es necesario que el paciente se posicione.
Quizá te interese
IMO Grupo Miranza Barcelona
Josep María Lladó, 3
08035 Barcelona
Tel: 934 000 700
E-mail: informacion@imo.es
Ver mapa en Google Maps
En coche
Coordenadas navegador GPS:
41º 24’ 38” N – 02º 07’ 29” E
Salida 7 de la Ronda de Dalt (lado montaña). La clínica cuenta con un aparcamiento de más de 200 plazas.
En bus
Autobús H2: Rotonda de Bellesguard, parada 1540
Autobús 196: Josep Maria Lladó-Bellesguard, parada 3191
Autobuses H2, 123, 196: Ronda de Dalt – Bellesguard, parada 0071
Cómo llegar a IMO desde:
IMO Grupo Miranza Madrid
C/ Valle de Pinares Llanos, 3
28035 Madrid
Tel: 910 783 783
E-mail: administracion.madrid@imo.es
Transporte público
Metro Lacoma (línea 7)
Autobuses:
- Líneas 49 y 64, parada “Senda del Infante”
- Línea N21, parada «Metro Lacoma»
Horarios
Atención al paciente: de lunes a viernes, de 8 h a 21 h
IMO Grupo Miranza Andorra
Av. de les Nacions Unides, 17
AD700 Escaldes-Engordany, Andorra
Tel: (+376) 88 55 44
Ver mapa en Google Maps