They affect less than
1
in every 2,000 people
More than
250
related genes are known
What are retinal dystrophies?
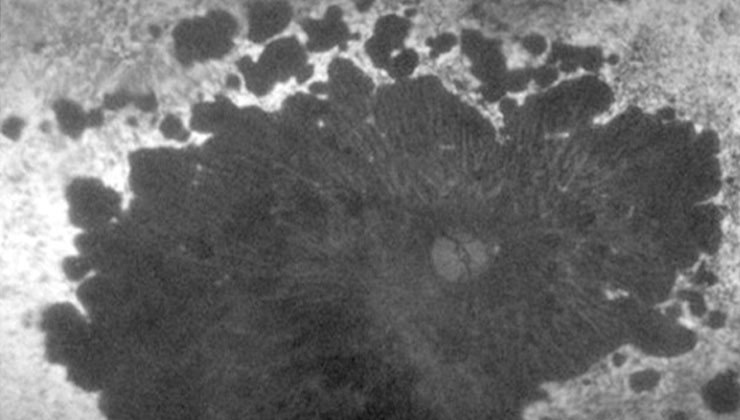
Retinal dystrophies are a heterogeneous group of hereditary diseases that cause progressive and severe loss of vision by altering the anatomy and/or function of the retina. There is currently no cure, but research is being carried out to find ways of treating it in the coming years with gene and cell therapies.
These conditions can cause damage to the photoreceptor cells, either, predominantly, the cones (responsible for detailed vision and colour), rods (responsible for night and peripheral vision) or both at the same time. This is the case of Stargardt disease, retinitis pigmentosa or cone-rod dystrophy, respectively.
There are also certain hereditary dystrophies, such as juvenile retinoschisis, familial exudative vitreoretinopathy and Stickler syndrome, in which vitreous and retinal abnormalities occur. In others, such as choroideremia, the underlying problem is in the choroid, the layer below the retina.
The majority of retinal dystrophies are diseases located exclusively in the eye, but can sometimes be associated with extraocular manifestations (Usher syndrome, Bardet-Biedl syndrome), in which case they are referred to as syndromic retinal dystrophies.
Due to their low prevalence, retinal dystrophies are considered minority or ‘rare’ diseases (affecting less than 1 in 2,000 people).
What causes them?
Retinal dystrophies are genetic in origin, meaning that they can be transmitted from generation to generation through different types of inheritance.
- Dominant inheritance: all generations of a family are usually affected because the carriers of the mutation responsible for the disease have the condition. This is transmitted to approximately 50% of descendants, such as in Best disease.
- Recessive inheritance: only family members in one generation are usually affected because the carriers of one mutation are healthy, while the carriers of two mutations in the same gene develop the disease. This is the case of Stargardt disease.
- Inheritance linked to the X chromosome: only men in the family suffer from the condition, but women can be carriers of the mutation and transmit the disease to their sons with a 50% probability. This group includes choroideremia, among others.
Retinitis pigmentosa, the most common retinal dystrophy, is an example of a condition that can be transmitted through the three inheritance patterns mentioned, depending on the gene involved.
The genetics of retinal dystrophies is complex: the same condition can be caused by several genes and, at the same time, the same gene can be related to different diseases. To date, more than 250 genes associated with retinal dystrophies have been described, but it is believed that there are still many to be identified.
How can they be prevented?
The genetic information contained in the DNA of every person determines whether a retinal dystrophy will develop, and, consequently, these hereditary conditions cannot be prevented. One aspect, however, that can make a difference is early detection by specialist ophthalmologists through a full eye check-up, including a good anamnesis and a thorough examination of the fundus, as well as the use of complementary techniques such as OCT, autofluorescence and electrophysiological tests.
Retinal dystrophies are often not evident until their advanced stages, when their symptoms become apparent. In order to anticipate the disease and predict its evolution, it is highly useful to know the molecular cause that triggers it in each patient by means of genetic diagnosis. This enables the clinical diagnosis to be confirmed, the inheritance pattern to be identified and genetic counselling to be offered to family members, who are alerted to the probability of the condition being transmitted and the possibility of them being carriers.
Genetic diagnosis also opens up the possibility of preventing, stopping or reversing the future loss of vision of affected patients through the development of new individualised therapies that are currently being researched.
Symptoms
Symptoms, as well as the speed of evolution, vary depending on the type of retinal dystrophy and each patient. The most common, however, are:
Loss of visual acuity: difficulty in making out fine detail and performing precision tasks.
Reduced field of vision: tunnel vision or the appearance of blind spots (scotomas).
Night blindness: problems seeing at night or in dark environments and poor adaptation to low-light situations.
Photophobia and glare: particular discomfort caused by light and seeing flashes or flickering light in high-light conditions. Moving from bright to dark environments –and vice versa– can also cause problems.
Other possible signs include distorted perception of objects (metamorphopsia) and alteration in the perception of colours (dyschromatopsia).
Associated treatments
Retinal dystrophies currently have no cure because of the difficulty of regenerating the affected retinal cells. Future treatment of these conditions involves designing and applying new gene and cell therapies to enable vision to be restored or its loss to be slowed down. Currently, significant steps are being taken in this line of research and the IMO’s Department of Genetics is also participating through several projects promoted by the IMO Foundation.
To prepare candidates for these gene therapies, it is essential to provide them with genetic diagnosis, an aspect in which the IMO is a pioneer.









FAQs
There are several eye diseases that are linked to chromosomal inheritance and even the genetic defect is determined. Diseases of all parts of the eye can be inherited. The most common is retinitis pigmentosa.
If there is no gas or silicone oil, the patient can sleep in any position. If there is no covering element (gas or silicone oil), the patient’s position is unimportant.
You may be interested in
IMO Institute of Ocular Microsurgery
Josep María Lladó, 3
08035 Barcelona
Phone: (+34) 934 000 700
E-mail: international@imo.es
See map on Google Maps
By car
GPS navigator coordinates:
41º 24’ 38” N – 02º 07’ 29” E
Exit 7 of the Ronda de Dalt (mountain side). The clinic has a car park with more than 200 parking spaces.
By bus
Autobus H2: Rotonda de Bellesguard, parada 1540
Autobus 196: Josep Maria Lladó-Bellesguard, parada 3191
Autobuses H2, 123, 196: Ronda de Dalt – Bellesguard, parada 0071
How to arrive at IMO from:
IMO Madrid
C/ Valle de Pinares Llanos, 3
28035 Madrid
Phone: (+34) 910 783 783
E-mail: administracion.madrid@imo.es
Public transport
Metro Lacoma (líne 7)
Autobuses:
- Lines 49 & 64, stop “Senda del Infante”
- Line N21, stop “Metro Lacoma”
Timetables
Patient care:
Monday to Friday, 8 a.m. to 9 p.m.
IMO Andorra
Av. de les Nacions Unides, 17
AD700 Escaldes-Engordany, Andorra
Phone:(+376) 88 55 44
See map in Google Maps